Surani Gunasekera from Cohort 3 has had her first paper published, you can read the full paper here. Lewis Hart from Cohort 1 is also a co-author on the paper!
Here is what Surani has to say about it.
Layered transition metal dichalcogenides are a group of materials that can be thinned down to atomic thicknesses. This is possible because the bulk material is composed of weakly coupled sandwich-like layers of metal and chalcogen atoms which can be relatively easily separated. In most of the transition metal dichalcogenides, atoms are arranged in one of two ways (lattices), reminiscent of tiling a plane with regular hexagons. However, in several of them, competition between those two structures results in more complicated, distorted atomic arrangements.
In our paper, we have studied the electronic band structure (the relation between momentum and energy of electrons) of two such distorted, low-symmetry materials: rhenium diselenide and rhenium disulphide. They are interesting because it has been suggested that the low symmetry makes their layers even less interacting than in other layered materials, making them “bulk two-dimensional materials”. At the same time, their complicated atomic structure makes predictions regarding their physical properties (for example, light with which frequency will be absorbed by the material and to what extent) difficult.
We have used a set of computational methods referred to jointly as density functional theory to study the electronic states in bulk ReSe2 and ReS2. In particular, in contrast to the usual approach, we investigated the whole range of the energy-momentum relation and not just the high-symmetry directions. This allowed us to identify momenta which host the top of the valence and bottom of the conduction band – states that set the electronic bandgap (range of energies forbidden for electrons) of a material. It turns out that in rhenium transition metal dichalcogenides these states can be easily missed because they are not along any of the special directions. Furthermore, our calculations indicate that several regions in the momentum space host similar local band gaps, which may be the reason for the measurements of optical reflection or absorption having difficulty in identifying the nature of the band gap in these materials.
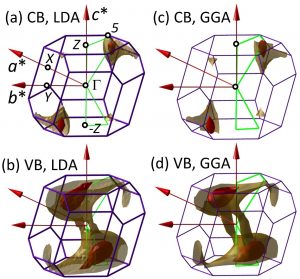
Constant energy surfaces in the Brillouin zone of ReS2. Calculations using LDA: (a) contours in the conduction band (CB) at energies of 70 meV (yellow) and 20 meV (red) above the CB minimum and (b) contours in the valence band (VB) at energies of 310 meV (yellow) and 70 meV (red) below the VB maximum.
https://link.springer.com/article/10.1007/s11664-018-6239-0;